
Publication date: April 2014 / Download as PDF
Author: Ibtissem Triki
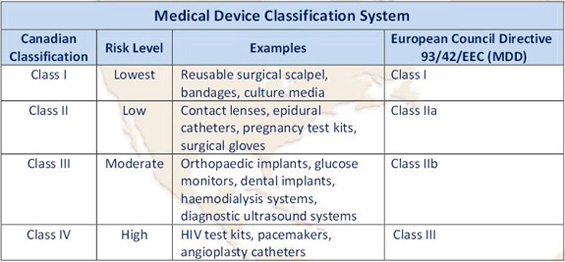
Canadian Medical Device Classifications
Investigational Testing in Human Clinical Trials
In Canada, Sponsors or Manufacturers should submit an Investigational Testing Application “ITA” to use unapproved Class II, III, and IV devices in clinical trials to the Device Evaluation Division of the Medical Devices office of Health Canada. Clinical trials for medical devices are not classified by phased development as with drugs (i.e., Phase I-IV clinical trials) and as for any standard clinical study in a human, the sponsor should still:
- Protect the rights, safety and well-being of human subjects;
- Ensure that the study is conducted with integrity, and that the best interests of the patients are always considered;
- Ensure the scientific conduct of the clinical investigation and the credibility of the results.
Devices must be labeled “for investigational use only”, and a Class I medical device does not require investigational testing authorization.
The Health Canada application should include:
- A protocol which details the objectives, benefits, risks, methods, and conditions for the trial to function;
- Research Ethics Board Attestation (REBA);
- Clinical Trial Site Information Form (CTSI);
- Investigational Testing Application;
- Qualified Investigator Undertaking (QIU)
In addition, based on the Special Access Program, a medical device that is not yet tested or licensed in Canada can be used in emergency cases where conventional or available treatments have failed. In such a case, health care professionals can apply for authorization to use medical devices through the Therapeutic Products Directorate to assess whether potential risks of using the device outweigh potential benefits.
Investigational Testing Application “ITA”
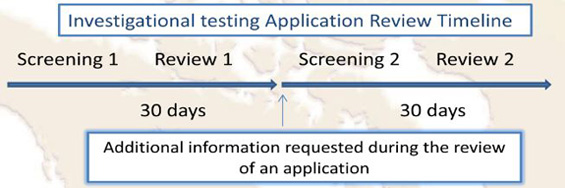
When applying for an authorization (to perform a clinical study to assess the safety and performance of a new medical device) to Health Canada, you need to submit an ITA according to the Canadian health program governing the use of Class II, III, and IV medical devices. The requirements for this application are summarized in the table below:

As per the Medical Devices Division of Health Canada, the target review time for a Class II, III, and IV medical device ITA is a total of 30 calendar days. First, the application will go through a screening process, and if it is accepted for review, a screening acceptance letter will be issued.
After approval of an ITA, a “No Objection Letter” (NOL) is sent to the Sponsor indicating Health Canada’s agreement on conducting the study. However, Health Canada requires notification of ethics clearance for the study through a Research Ethics Board prior to issuing a No Objection letter.
If Unlicensed Devices are to be used in conjunction with a Drug in a clinical trial, the Sponsor should sign the application for both an ITA (Devices) and CTA (Drugs) that will be reviewed in parallel.
Challenges in Medical Device Clinical Development in Canada
The clinical development of a new medical device involves exploratory (pilot) and pivotal clinical studies as well as in vitro and in vivo pre-clinical investigations, if possible. Pilot studies are performed in the early phase of medical device development to capture preliminary safety and effectiveness information on the device before the pivotal study design, and are typically performed at one or two sites with less than 100 patients. Based on pilot study work, Pivotal studies are conducted to determine the safety and effectiveness of a device in a statistically justified number of subjects.
In Canada, there is no equivalent to the FDA 510(k) clearance that allows a medical device company to bypass the expensive and time consuming randomized clinical trial process. FDA 510(k) clearance requires that the sponsor demonstrate that there is a substantial equivalence to products already approved in teh US market and that the difference between the “new” and the existing device is acceptable for FDA clearance. Therefore, Health Canada’s requirements for class II, III and IV devices are higher than the standards employed in the US. In addition, over the last five years, the Canadian regulation requirements have become more stringent, which has resulted in significant delays for starting up Clinical studies for a medical device. This, along with the continued increase of clinical research costs, has forced Sponsors or Manufacturers to re-evaluate clinical development approaches for medical devices.
Monitoring Medical Device Trials
Clinical site, investigator, IRB/EC requirements cannot be less stringent than ISO14155-2011, if the standard is part of the requirements for the study. The new ISO14155-2011 “Clinical investigation of medical devices for human subjects-Good clinical practice” is more harmonized with ICH GCP but adapted for medical device trials and has been well received as a global standard for medical device trials than the two earlier versions of the document ISO14155-1996, and ISO 14155-2003. Other than focusing more on Project Management, GCP requirements and documentation, this complete revised version emphasizes on monitoring activities, and the monitor’s role in supporting compliance. Additionally, this version also regulates the electronic data capture systems used in medical device studies that are a critical step in central monitoring activities and part of the new FDA guidelines regarding the Risk-based monitoring approach. This methodology uses adaptive monitoring based on risk assessments that increase Clinical Research Associate (CRA) efficiency and productivity combined to data quality and subject safety.

In conclusion, Vantage BioTrials has a deep understanding of the challenges faced in clinical development for medical devices in Canada. Our team can provide experienced professionals to meet your clinical development needs for both pilot and pivotal studies using efficient study management strategies that can reduce costs and shorten time lines by:
Developing a well-designed protocol and case report forms (CRFs) to ensure high quality data and efficient monitoring;
- Site identification and feasibility analysis;
- Support Services to meet ISO 14971-2012 “Application of risk management to medical devices” requirement; management procedures and practices to analyze, evaluate, control, and monitor risk relating to the safety of a medical device throughout the design, development and product lifecycle;
- Investigational Testing Application to Health Canada, IRB/EC submission, Labeling assistance, and other global requirements. Efficiently managing study start up, and execution in accordance with ICH GCP & ISO 14155-2011, and Canadian Medical device regulations;
- Risk analysis/mitigation and risk-based monitoring approach.
About the Author
Ibtissem Triki is a Clinical Research Specialist at Vantage BioTrials, Inc. With over 14 years of Pharmaceutical & Academic experience, Ibtissem brings to Vantage BioTrials a wealth of clinical research knowledge. She has a strong understanding and experience with the conduct and implementation of clinical research and medical device studies and in accordance to regulatory and agency requirements.
References
- ISO 14155:2011, Clinical investigation of medical devices for human subjects-Good clinical practice, published 1 February2011: http://www.iso.org/iso/catalogue_detail?csnumber=45557
- ISO 14971:2007, Medical devices, Application of risk management to medical device: http://www.iso.org/iso/home/store/catalogue_tc/catalogue_detail.htm?csnumber=38193
- WMDO, Webinar on “The Hidden Challenges of the New ISO 14155 Requirements, 2 February 2011: http://www.wmdo.org/article-detail.aspx?id=12
- FDA Guidance for Industry, Oversight of Clinical. Investigations-A risk-Based Approach to Monitoring (August 2013): http://www.fda.gov/downloads/Drugs/…/Guidances/UCM269919.pdf
- Health Canada Medical Device Regulations (Last amended on December 16, 2011): http://laws-lois.justice.gc.ca/PDF/SOR-98-282.pdf
- Preparation of an Application for Investigational Testing-Medical Devices (Feb 1999). Therapeutic Products Program Guidance Document: http://www.hc-sc.gc.ca/–demande/guide-ld/md_gd_ita_im_ld_aeeeng.php
- FDA Premarket Notification (510k): http://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketnotification510k/default.htm